18.9 Распространённость, получение и соединения кислорода (Occurrence, Preparation, and Compounds of Oxygen)¶
Цели обучения
К концу этого раздела вы сможете:
- описывать свойства, получение и соединения кислорода;
- описывать получение, свойства и применение некоторых типичных оксидов, пероксидов и гидроксидов металлов.
Кислород — самый распространённый элемент земной коры. Поверхностный слой Земли составляют кора, атмосфера и гидросфера. Около 50 % массы земной коры приходится на кислород (в соединениях с другими элементами, главным образом с кремнием). В воздухе кислород присутствует в виде молекул \(\ce{O2}\) и в небольшом количестве — в виде молекул \(\ce{O3}\) (озона); на его долю приходится около 20 % массы воздуха. Около 89 % массы воды составляет связанный кислород. В соединениях с углеродом, водородом и азотом кислород входит в значительной доле в состав растений и животных.
При обычных температурах кислород — бесцветный газ без запаха и вкуса. Он немного плотнее воздуха. Хотя кислород лишь немного растворим в воде (49 мл газа растворяется в 1 л при стандартных температуре и давлении), его растворимость очень важна для водных организмов.
Бо́льшую часть кислорода в промышленных масштабах получают из воздуха, а остальное — электролизом воды. Отделение кислорода от воздуха начинают с того, что воздух охлаждают и сжимают до его сжижения. При нагревании жидкого воздуха кислород, имеющий более высокую температуру кипения (\(90\ \text{К}\)), отделяется от азота, температура кипения которого ниже (\(77\ \text{К}\)). Одновременно по разности температур кипения можно выделить и остальные компоненты воздуха.
Кислород необходим для процессов горения, в том числе для сжигания топлива. Растения и животные используют кислород воздуха при дыхании. Подача обогащённого кислородом воздуха — важная медицинская процедура для пациентов, испытывающих недостаток кислорода вследствие шока, пневмонии или другой болезни.
В химической промышленности кислород используют для окисления многих веществ. Значительная доля производимого кислорода идёт на удаление углерода из железа при выплавке стали. Большие количества чистого кислорода необходимы в металлообработке, а также при резке и сварке металлов водородно-кислородным и кислородно-ацетиленовым пламенем.
Жидкий кислород важен для космической отрасли. Он служит окислителем в ракетных двигателях, а также источником газообразного кислорода для систем жизнеобеспечения в космосе.
Как известно, кислород очень важен для жизни. Энергия, необходимая для поддержания нормальных функций организма у человека и других живых существ, выделяется при медленном окислении химических соединений. Конечным окислителем в этих реакциях служит кислород. У человека кислород проходит из лёгких в кровь, где соединяется с гемоглобином, образуя оксигемоглобин. В таком виде кровь переносит кислород к тканям, где он передаётся клеткам и расходуется как химический окислитель; конечными продуктами становятся углекислый газ и вода. Кровь по венам уносит углекислый газ в лёгкие, где отдаёт его и забирает новую порцию кислорода. Переваривание и усвоение пищи восполняют вещества, израсходованные в результате окисления в организме; высвобождаемая при этом энергия равна той, которая выделилась бы при сжигании пищи вне организма.
Зелёные растения непрерывно пополняют запас кислорода в атмосфере в процессе фотосинтеза (photosynthesis). Продукты фотосинтеза могут различаться, но в общем виде процесс превращает углекислый газ и воду в глюкозу (сахар) и кислород за счёт энергии света:
Таким образом, кислород, ушедший в виде углекислого газа и воды в результате метаболизма у растений и животных, возвращается в атмосферу через фотосинтез.
Если пропускать сухой кислород между двумя электрически заряженными пластинами, образуется озон (ozone, \(\ce{O3}\), показан на рис. 18.42) — аллотроп кислорода с характерным запахом. Образование озона из кислорода — эндотермическая реакция, энергия для которой поступает от электрического разряда, тепла или ультрафиолетового излучения:
Резкий запах, возникающий при искрении электрооборудования, частично обусловлен озоном.
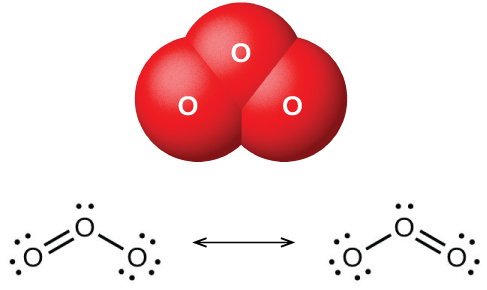
Рис. 18.42. Уголковая молекула озона (\(\ce{O3}\)) и резонансные структуры, необходимые для описания её связей.
Озон образуется естественным путём в верхних слоях атмосферы под действием солнечного ультрафиолетового излучения на находящийся там кислород. Бо́льшая часть атмосферного озона сосредоточена в стратосфере — слое атмосферы, простирающемся примерно от 10 до 50 километров над поверхностью Земли. Этот озон служит барьером для опасного ультрафиолетового излучения Солнца, поглощая его в реакции химического разложения:
Реакционноспособные атомы кислорода вновь соединяются с молекулярным кислородом, замыкая озоновый цикл. Наличие стратосферного озона снижает частоту рака кожи и других вредных последствий ультрафиолетового излучения. Убедительно показано, что хлорфторуглероды (ХФУ, известные под торговым названием фреоны), применявшиеся в качестве пропеллентов в аэрозольных баллончиках и как хладагенты, вызывали уменьшение содержания озона в стратосфере. Это происходит потому, что ультрафиолетовое излучение разлагает и сами ХФУ, образуя атомарный хлор. Атомы хлора реагируют с молекулами озона, что в итоге уменьшает количество \(\ce{O3}\) в стратосфере. Подробно этот процесс рассмотрен в главе по химической кинетике. В мире прилагают усилия для сокращения промышленного использования ХФУ, и озоновая дыра уже начала сокращаться по мере уменьшения концентрации атомарного хлора в атмосфере. Если стратосферный озон защищает нас, то озон в тропосфере — это проблема: он является токсичным компонентом фотохимического смога.
Применение озона основано на его реакционной способности по отношению к другим веществам. Его можно использовать как отбеливатель для масел, восков, тканей и крахмала: озон окисляет окрашенные соединения в этих веществах до бесцветных. Озон — альтернатива хлору при обеззараживании воды.
Реакции¶
Элементный кислород — сильный окислитель. Он реагирует с большинством других элементов и со многими соединениями.
Реакция с элементами¶
Кислород при комнатной или повышенной температуре непосредственно реагирует со всеми элементами, кроме благородных газов, галогенов и нескольких малореакционноспособных переходных металлов второго и третьего рядов (тех, у которых восстановительные потенциалы выше, чем у меди). Пример реакции кислорода с железом — ржавление. Более активные металлы образуют пероксиды или супероксиды, менее активные металлы и неметаллы — оксиды. Два примера таких реакций:
Оксиды галогенов, по крайней мере одного из благородных газов и металлов с восстановительными потенциалами выше, чем у меди, прямым взаимодействием соответствующих элементов с кислородом не получаются.
Реакция с соединениями¶
Элементный кислород реагирует и с некоторыми соединениями. Если в данном соединении какой-либо из элементов способен окисляться, дальнейшее окисление кислородом возможно. Например, в сероводороде \(\ce{H2S}\) сера имеет степень окисления \(-2\). Поскольку сера здесь не находится в максимальной степени окисления, можно ожидать, что \(\ce{H2S}\) реагирует с кислородом. Так и происходит — образуются вода и диоксид серы:
Можно окислить и такие оксиды, как \(\ce{CO}\) и \(\ce{P4O6}\), содержащие элемент в более низкой степени окисления. Той лёгкости, с которой элементный кислород присоединяет электроны, соответствует трудность отрыва электронов от кислорода в большинстве оксидов. Среди элементов лишь очень активный фтор способен окислить оксиды до газообразного кислорода.
Оксиды, пероксиды и гидроксиды¶
Соединения типичных металлов с кислородом распадаются на три класса: (1) оксиды (oxides), содержащие оксидные ионы \(\ce{O^{2-}}\); (2) пероксиды (peroxides), содержащие пероксид-ионы \(\ce{O2^{2-}}\) с ковалентной одинарной связью кислород–кислород, и очень небольшое число супероксидов (superoxides), содержащих супероксид-ион \(\ce{O2-}\) с ковалентной кислород–кислородной связью порядка \(1\tfrac{1}{2}\). Кроме того, выделяют (3) гидроксиды (hydroxides), содержащие гидроксид-ион \(\ce{OH-}\). Оксиды образуют все типичные металлы. Некоторые металлы группы 2 образуют и пероксиды \(\ce{MO2}\), а металлы группы 1 — пероксиды \(\ce{M2O2}\) и супероксиды \(\ce{MO2}\).
Оксиды¶
Оксиды большинства типичных металлов можно получить нагреванием соответствующих гидроксидов (тогда образуются оксид и газообразная вода) или карбонатов (образуются оксид и газообразный \(\ce{CO2}\)). Примеры таких реакций:
Однако соли щелочных металлов обычно очень устойчивы и при нагревании не разлагаются легко. Оксиды щелочных металлов получают окислительно-восстановительными реакциями при нагревании нитратов или гидроксидов с самим металлом. Примеры реакций:
За исключением оксида ртути(II), оксиды металлов групп 2–15 можно получить, сжигая соответствующий металл на воздухе. Самый тяжёлый член каждой группы — тот, у которого наиболее выражен эффект инертной пары, — образует оксид, в котором степень окисления металла на две единицы меньше групповой (эффект инертной пары). Так, при сжигании таллия, свинца и висмута образуются соответственно \(\ce{Tl2O}\), \(\ce{PbO}\) и \(\ce{Bi2O3}\). Оксиды более лёгких членов каждой группы отвечают групповой степени окисления; например, при сжигании олова образуется \(\ce{SnO2}\). Оксид ртути(II), \(\ce{HgO}\), медленно образуется при нагревании ртути ниже \(500\ \text{°C}\) и разлагается при более высоких температурах.
Сжигание представителей групп 1 и 2 на воздухе непригодно для получения оксидов этих элементов: эти металлы достаточно реакционноспособны, чтобы соединяться и с азотом воздуха, поэтому образуются смеси оксидов и ионных нитридов. Некоторые из них при нагревании на воздухе образуют ещё и пероксиды или супероксиды.
Ионные оксиды все содержат оксидный ион — очень сильный акцептор иона водорода. За исключением очень малорастворимого оксида алюминия \(\ce{Al2O3}\), оксида олова(IV) \(\ce{SnO2}\) и оксида свинца(IV) \(\ce{PbO2}\), оксиды типичных металлов реагируют с кислотами с образованием солей. Примеры таких реакций:
Оксиды металлов групп 1 и 2, а также оксид таллия(I) реагируют с водой, образуя гидроксиды. Примеры таких реакций:
Оксиды щелочных металлов промышленного значения почти не имеют — в отличие от оксида магния, оксида кальция и оксида алюминия. Оксид магния важен в производстве огнеупорного кирпича, тиглей, футеровки печей и теплоизоляции — там, где требуются химическая и термическая устойчивость. Оксид кальция, который в промышленной практике называют негашёной известью (quicklime) или просто известью (lime), очень реакционноспособен, и его основные применения именно с этим связаны. Чистый оксид кальция, нагретый до высокой температуры, испускает интенсивный белый свет (см. рис. 18.43). Раскалённые газовым пламенем бруски оксида кальция служили сценическим освещением в театрах до появления электрического света. Отсюда выражение «в свете рампы» (английская идиома in the limelight, буквально «в известковом свете»).
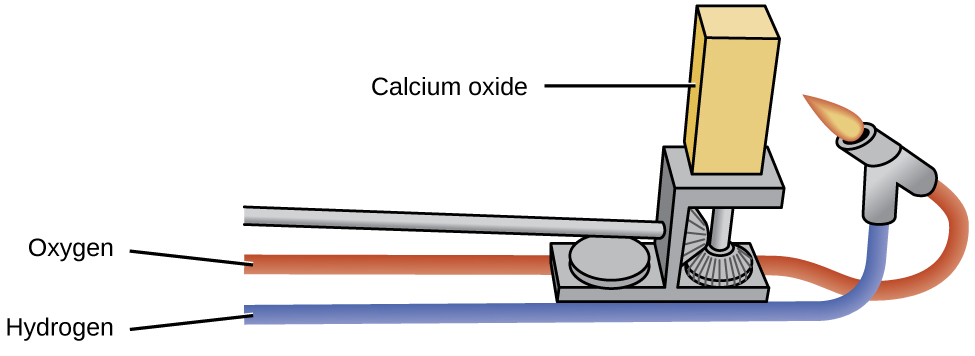
Рис. 18.43. Оксид кальция имеет множество промышленных применений. При нагревании до высокой температуры он испускает интенсивный белый свет.
Оксид кальция и гидроксид кальция — дешёвые основания, широко используемые в химической технологии, хотя бо́льшая часть полезных продуктов, получаемых из них, кальция не содержит. Оксид кальция \(\ce{CaO}\) получают нагреванием карбоната кальция \(\ce{CaCO3}\), который доступен и недорог в виде известняка или раковин устриц:
Хотя эта реакция разложения обратима, можно получить 100 %-ный выход \(\ce{CaO}\), отводя \(\ce{CO2}\). Гидроксид кальция получают привычной кислотно-основной реакцией растворимого оксида металла с водой:
И \(\ce{CaO}\), и \(\ce{Ca(OH)2}\) полезны как основания: они принимают протоны и нейтрализуют кислоты.
Алюминий-оксид (alumina, \(\ce{Al2O3}\)) встречается в природе как минерал корунд (corundum) — очень твёрдое вещество, используемое как абразив для шлифовки и полировки. Корунд важен в ювелирном деле как рубин и сапфир. Окраска рубина обусловлена небольшой примесью хрома; другие примеси дают разнообразие цветов сапфира. Искусственные рубины и сапфиры в наши дни получают, расплавляя оксид алюминия (температура плавления \(2050\ \text{°C}\)) с небольшим количеством оксидов для придания нужной окраски и кристаллизуя расплав так, чтобы вырастить крупные кристаллы. В рубиновых лазерах используют синтетические кристаллы рубина.
Оксид цинка \(\ce{ZnO}\) когда-то был полезным белым пигментом, однако загрязнения окружающей среды приводят к его обесцвечиванию. Этот оксид важен также в производстве автомобильных шин и других резиновых изделий, а также при изготовлении лечебных мазей. Например, солнцезащитные средства на основе оксида цинка (рис. 18.44) защищают кожу от ожогов. Оксид цинка в таких средствах содержится в виде очень мелких зёрен, известных как наночастицы (nanoparticles). Диоксид свинца \(\ce{PbO2}\) — компонент заряженных свинцово-кислотных аккумуляторов. Свинец(IV) стремится перейти в более устойчивый ион свинца(II), присоединяя два электрона, поэтому диоксид свинца — сильный окислитель.
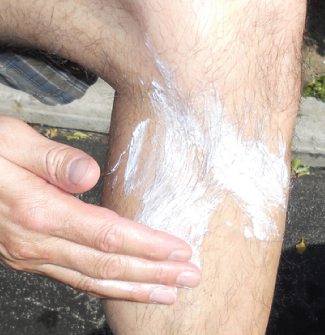
Рис. 18.44. Оксид цинка защищает открытую кожу от солнечных ожогов. (Источник: модификация работы «osseous» / Flickr.)
Пероксиды и супероксиды¶
Пероксиды и супероксиды — сильные окислители и важны в химических процессах. Пероксид водорода \(\ce{H2O2}\), получаемый из пероксидов металлов, — важный отбеливатель и дезинфицирующее средство. Пероксиды и супероксиды образуются при взаимодействии металлов или их оксидов из групп 1 и 2 с чистым кислородом при повышенных температурах. Пероксид натрия и пероксиды кальция, стронция и бария получают нагреванием соответствующего металла или его оксида в чистом кислороде:
Пероксиды калия, рубидия и цезия можно получить нагреванием металла или его оксида в строго отмеренном количестве кислорода:
С избытком кислорода образуются супероксиды \(\ce{KO2}\), \(\ce{RbO2}\) и \(\ce{CsO2}\). Например:
Устойчивость пероксидов и супероксидов щелочных металлов возрастает с увеличением размера катиона.
Гидроксиды¶
Гидроксиды — соединения, содержащие ион \(\ce{OH-}\). Их можно получить двумя общими способами. Растворимые гидроксиды металлов образуются при реакции металла или оксида металла с водой. Нерастворимые гидроксиды металлов образуются при смешивании раствора растворимой соли металла с раствором, содержащим гидроксид-ионы.
За исключением бериллия и магния, металлы групп 1 и 2 реагируют с водой, образуя гидроксиды и газообразный водород. Примеры таких реакций:
Однако эти реакции могут быть бурными и опасными; поэтому растворимые гидроксиды металлов предпочтительнее получать реакцией соответствующего оксида с водой:
Большинство оксидов металлов — ангидриды оснований (base anhydrides). Это очевидно для растворимых оксидов, так как они образуют гидроксиды металлов. Большинство остальных оксидов металлов нерастворимы и не образуют гидроксидов в воде; тем не менее они тоже являются ангидридами оснований, потому что реагируют с кислотами.
Нерастворимые гидроксиды бериллия, магния и других типичных металлов можно получить, добавляя гидроксид натрия к раствору соответствующей соли металла. Краткие ионные уравнения реакций для соли магния, соли алюминия и соли цинка:
При получении гидроксидов алюминия, галлия, цинка и олова(II) следует избегать избытка гидроксида, иначе осадки растворяются с образованием соответствующих комплексных ионов: \(\ce{Al(OH)4-}\), \(\ce{Ga(OH)4-}\), \(\ce{Zn(OH)4^{2-}}\) и \(\ce{Sn(OH)3-}\) (см. рис. 18.45). Для нашей главы важно, что эти комплексные ионы образуются по кислотно-основной реакции Льюиса, в которой металл выступает кислотой Льюиса.

Рис. 18.45. (a) При смешивании растворов \(\ce{NaOH}\) и \(\ce{Zn(NO3)2}\) образуется белый осадок \(\ce{Zn(OH)2}\). (b) Добавление избытка \(\ce{NaOH}\) приводит к растворению осадка. (Источник: модификация работы Mark Ott.)
Промышленность использует большие количества гидроксида натрия как дешёвого сильного основания. Исходным веществом для производства \(\ce{NaOH}\) служит хлорид натрия, поскольку \(\ce{NaCl}\) дешевле оксида. Гидроксид натрия входит в десятку химических веществ, производимых в США в наибольших количествах, причём почти весь его выпуск получают электролизом растворов хлорида натрия. Этот процесс — хлор-щелочной процесс (chlor-alkali process), он же является основным способом получения хлора.
Гидроксид натрия — ионное соединение, плавится без разложения. Очень хорошо растворим в воде, при этом выделяется значительное количество теплоты и образуются сильно щелочные растворы: при \(25\ \text{°C}\) 40 граммов гидроксида натрия растворяется всего лишь в 60 граммах воды. Гидроксид натрия применяют для получения других соединений натрия и нейтрализации кислых растворов при производстве иных химических продуктов — нефтехимии и полимеров.
Многие применения гидроксидов связаны с нейтрализацией кислот (например, антацида, показанного на рис. 18.46) и с получением оксидов путём термического разложения. Водная суспензия гидроксида магния — это антацид «магнезиальное молочко». Благодаря доступности (его получают реакцией воды с оксидом кальция, который, в свою очередь, образуется при разложении известняка \(\ce{CaCO3}\)), низкой стоимости и высокой активности гидроксид кальция широко используется в технических применениях, где требуется дешёвое сильное основание. Реакцией гидроксидов с подходящими кислотами получают также соли.

Рис. 18.46. Карбонат кальция, \(\ce{CaCO3}\), можно принимать как антацид для нейтрализации действия кислоты в желудке. (Источник: «Midnightcomm» / Wikimedia Commons.)
Химия в повседневной жизни. Хлор-щелочной процесс
Хотя хлор и гидроксид натрия химически очень разные, между ними есть связь, потому что существует важный электрохимический процесс, в котором эти два вещества образуются одновременно. Этот процесс, известный как хлор-щелочной процесс (chlor-alkali process), использует хлорид натрия, крупные залежи которого есть во многих регионах мира. Это электрохимический процесс окисления хлорид-иона до хлора с одновременным получением гидроксида натрия.
При пропускании постоянного электрического тока через раствор \(\ce{NaCl}\) хлорид-ионы мигрируют к положительному электроду, где окисляются до газообразного хлора, отдавая электрон электроду:
Образовавшиеся электроны проходят по внешней электрической цепи к отрицательному электроду. Хотя положительные ионы натрия движутся к этому отрицательному электроду, металлический натрий не образуется, так как при заданных условиях ионы натрия слишком трудно восстановить. (Напомним, что металлический натрий настолько активен, что реагирует с водой, и даже если бы он образовался, то немедленно взаимодействовал бы с водой, снова давая ионы натрия.) Вместо этого молекулы воды принимают электроны от электрода и восстанавливаются с образованием газообразного водорода и гидроксид-ионов:
Суммарный итог — превращение водного раствора \(\ce{NaCl}\) в водный раствор \(\ce{NaOH}\), газообразный \(\ce{Cl2}\) и газообразный \(\ce{H2}\):
Кислородные соединения неметаллов¶
Большинство неметаллов реагирует с кислородом, образуя оксиды неметаллов. В зависимости от доступных для элемента степеней окисления могут образовываться разнообразные оксиды. Фтор соединяется с кислородом, образуя фториды, такие как \(\ce{OF2}\), в которых кислород имеет степень окисления \(+2\).
Кислородные соединения серы¶
Два распространённых оксида серы — диоксид серы \(\ce{SO2}\) и триоксид серы \(\ce{SO3}\). Запах горящей серы обусловлен диоксидом серы. Диоксид серы (рис. 18.47) встречается в вулканических газах и в атмосфере вблизи промышленных предприятий, сжигающих топливо с серосодержащими соединениями.
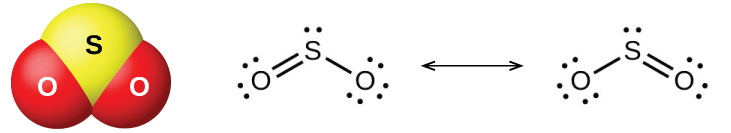
Рис. 18.47. Молекулярная структура (слева) и резонансные формы (справа) диоксида серы.
Промышленное производство диоксида серы основано либо на сжигании серы, либо на обжиге сульфидных руд — таких как \(\ce{ZnS}\), \(\ce{FeS2}\) и \(\ce{Cu2S}\) — на воздухе. (Обжиг, при котором образуется оксид металла, — первая стадия выделения многих металлов из их руд.) Удобный лабораторный способ получения диоксида серы — действие сильной кислоты на сульфиты, содержащие ион \(\ce{SO3^{2-}}\), или гидросульфиты, содержащие ион \(\ce{HSO3-}\). Сначала образуется сернистая кислота \(\ce{H2SO3}\), но она быстро разлагается на диоксид серы и воду. Диоксид серы образуется также при взаимодействии многих восстановителей с горячей концентрированной серной кислотой. Триоксид серы \(\ce{SO3}\) медленно образуется при совместном нагревании диоксида серы и кислорода; реакция экзотермическая:
Диоксид серы при комнатной температуре — газ, молекула \(\ce{SO2}\) уголковая. Триоксид серы плавится при \(17\ \text{°C}\) и кипит при \(43\ \text{°C}\). В парах его молекулы — отдельные \(\ce{SO3}\) (рис. 18.48), но в твёрдом состоянии \(\ce{SO3}\) существует в нескольких полимерных формах.
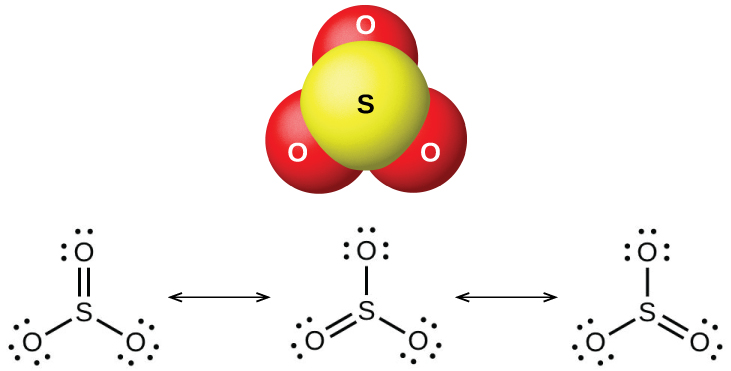
Рис. 18.48. Структура (вверху) триоксида серы в газовой фазе и его резонансные формы (внизу).
Оксиды серы реагируют как кислоты Льюиса со многими оксидами и гидроксидами в кислотно-основных реакциях Льюиса, образуя соответственно сульфиты или гидросульфиты, сульфаты или гидросульфаты.
Кислородные соединения галогенов¶
Галогены непосредственно с кислородом не реагируют, однако бинарные соединения кислород–галоген можно получить реакцией галогенов с кислородсодержащими веществами. Соединения кислорода с хлором, бромом и иодом — это оксиды, поскольку в них кислород является более электроотрицательным элементом. С другой стороны, соединения фтора с кислородом — фториды, потому что более электроотрицательным здесь является фтор.
Как класс эти оксиды чрезвычайно реакционноспособны и неустойчивы, их химия имеет мало практического значения. Дихлор-оксид (формально называемый монооксид дихлора) и диоксид хлора, оба показанные на рис. 18.49, — единственные коммерчески важные соединения. Они применяются как отбеливатели (для целлюлозы и муки) и для очистки воды.
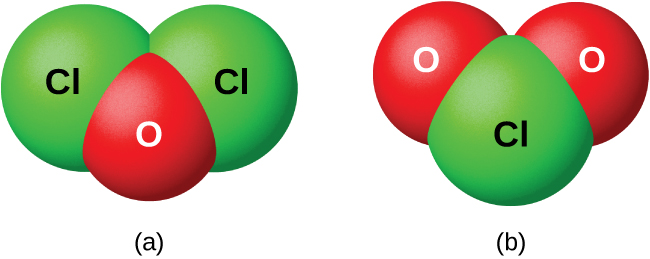
Рис. 18.49. Структуры молекул (a) \(\ce{Cl2O}\) и (b) \(\ce{ClO2}\).
Кислородсодержащие кислоты неметаллов и их соли¶
Оксиды неметаллов при взаимодействии с водой образуют кислоты; такие оксиды называют ангидридами кислот (acid anhydrides). Образующиеся оксоанионы могут давать соли с различными ионами металлов.
Оксикислоты азота и их соли¶
Пентаоксид азота \(\ce{N2O5}\) и \(\ce{NO2}\) реагируют с водой, образуя азотную кислоту \(\ce{HNO3}\). Алхимики ещё в восьмом веке знали азотную кислоту (рис. 18.50) под названием aqua fortis («крепкая вода»). Эта кислота была полезна для разделения золота и серебра, поскольку растворяет серебро, но не золото. Следы азотной кислоты появляются в атмосфере после грозовых разрядов, а её соли широко распространены в природе. Существуют огромные залежи чилийской селитры \(\ce{NaNO3}\) в пустынном регионе у границы Чили и Перу. Бенгальская селитра \(\ce{KNO3}\) встречается в Индии и других странах Дальнего Востока.
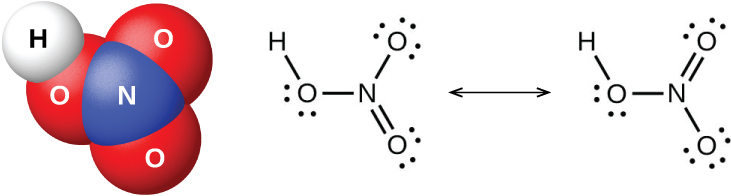
Рис. 18.50. Молекулярная структура (слева) азотной кислоты \(\ce{HNO3}\) и её резонансные формы (справа).
В лаборатории азотную кислоту можно получить нагреванием нитратной соли (например, нитрата натрия или калия) с концентрированной серной кислотой:
В промышленности азотную кислоту производят процессом Оствальда (Ostwald process). Он включает окисление аммиака до оксида азота \(\ce{NO}\), окисление \(\ce{NO}\) до диоксида азота \(\ce{NO2}\) и дальнейшее окисление и гидратацию \(\ce{NO2}\) до азотной кислоты:
или
Чистая азотная кислота — бесцветная жидкость. Однако она часто бывает жёлтой или коричневой из-за того, что при разложении кислоты образуется \(\ce{NO2}\). В водных растворах азотная кислота устойчива; коммерчески доступная концентрированная азотная кислота содержит 68 % кислоты. Это одновременно и сильный окислитель, и сильная кислота.
При действии азотной кислоты на металл редко выделяется \(\ce{H2}\) (за счёт восстановления \(\ce{H+}\)), да и то в малых количествах. Вместо этого происходит восстановление азота. Состав продуктов зависит от концентрации кислоты, активности металла и температуры. Обычно образуется смесь нитратов, оксидов азота и различных продуктов восстановления. Менее активные металлы — медь, серебро, свинец — восстанавливают концентрированную азотную кислоту преимущественно до диоксида азота. Разбавленная азотная кислота с медью даёт \(\ce{NO}\). В каждом случае при упаривании раствора кристаллизуются нитратные соли соответствующих металлов.
Неметаллы, такие как сера, углерод, иод и фосфор, окисляются концентрированной азотной кислотой до своих оксидов или оксикислот с одновременным выделением \(\ce{NO2}\):
Азотная кислота окисляет многие соединения; так, концентрированная азотная кислота легко окисляет хлороводородную кислоту до хлора и диоксида хлора. Смесь одного объёма концентрированной азотной кислоты и трёх объёмов концентрированной соляной кислоты — царская водка (aqua regia, «королевская вода») — энергично реагирует с металлами. Эта смесь особенно полезна для растворения золота, платины и других металлов, которые труднее окислить, чем водород. Упрощённое уравнение, описывающее действие царской водки на золото:
Дополнительно
Хотя золото обычно не реакционноспособно, можно посмотреть видеоролик, в котором сложная смесь соединений, присутствующих в царской водке, растворяет его.
Нитраты — соли азотной кислоты — образуются при взаимодействии металлов, оксидов, гидроксидов или карбонатов с азотной кислотой. Большинство нитратов растворимы в воде; собственно, одно из важных применений азотной кислоты — это получение растворимых нитратов металлов.
Азотная кислота широко применяется в лабораторной практике и в химической промышленности как сильная кислота и сильный окислитель. Она важна в производстве взрывчатых веществ, красителей, пластмасс и лекарств. Соли азотной кислоты (нитраты) ценны как удобрения. Дымный порох — смесь нитрата калия, серы и древесного угля.
Взаимодействие \(\ce{N2O3}\) с водой даёт бледно-голубой раствор азотистой кислоты \(\ce{HNO2}\). Однако \(\ce{HNO2}\) (рис. 18.51) проще получить, добавляя кислоту к раствору нитрита; азотистая кислота — слабая, поэтому нитрит-ион в водном растворе ведёт себя как основание:
Азотистая кислота очень неустойчива и существует только в растворе. При комнатной температуре она медленно (а при нагревании быстро) диспропорционирует на азотную кислоту и оксид азота(II). Азотистая кислота — активный окислитель по отношению к сильным восстановителям, а сильные окислители окисляют её до азотной кислоты.
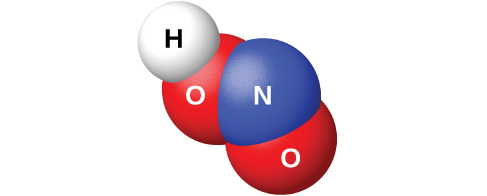
Рис. 18.51. Молекулярная структура молекулы азотистой кислоты \(\ce{HNO2}\).
Нитрит натрия \(\ce{NaNO2}\) — добавка к мясным продуктам, таким как сосиски и колбасные нарезки. Нитрит-ион выполняет две функции. Он ограничивает рост бактерий, вызывающих пищевые отравления, и удлиняет срок сохранения красного цвета мяса. Добавление нитрита натрия в мясные продукты вызывает споры, потому что азотистая кислота реагирует с некоторыми органическими соединениями, образуя класс веществ, известных как нитрозамины (nitrosamines). Нитрозамины вызывают рак у лабораторных животных. Это побудило Управление по санитарному надзору США (FDA) ограничить содержание \(\ce{NaNO2}\) в продуктах питания.
Нитриты значительно устойчивее, чем сама кислота, но, как и нитраты, могут взрываться. Нитриты, как и нитраты, растворимы в воде (\(\ce{AgNO2}\) лишь слегка растворим).
Кислородные кислоты фосфора и их соли¶
Чистая ортофосфорная кислота \(\ce{H3PO4}\) (рис. 18.52) образует бесцветные расплывающиеся (deliquescent) кристаллы, плавящиеся при \(42\ \text{°C}\). Обиходное название этого соединения — фосфорная кислота, в продаже она имеется в виде вязкого 82 %-ного раствора, известного как «сиропообразная фосфорная кислота». Одно из применений фосфорной кислоты — добавка во многие безалкогольные напитки.
Один из промышленных способов получения ортофосфорной кислоты — обработка фосфоритной породы концентрированной серной кислотой:
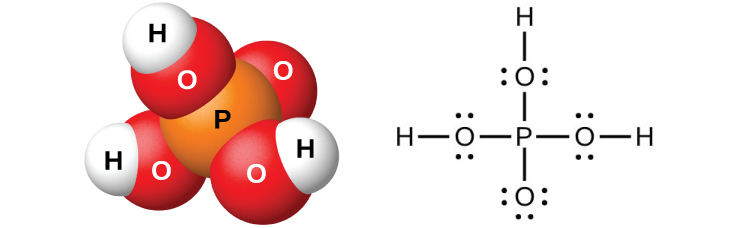
Рис. 18.52. В чистом виде ортофосфорная кислота \(\ce{H3PO4}\) бесцветна и имеет показанные молекулярную (слева) и льюисову (справа) структуры.
Разбавление продуктов водой с последующим отфильтровыванием сульфата кальция даёт разбавленный раствор кислоты, загрязнённый дигидрофосфатом кальция \(\ce{Ca(H2PO4)2}\) и другими соединениями, сопутствующими фосфоритной породе. Чистую ортофосфорную кислоту можно получить растворением \(\ce{P4O10}\) в воде.
Действие воды на \(\ce{P4O6}\), \(\ce{PCl3}\), \(\ce{PBr3}\) или \(\ce{PI3}\) даёт фосфористую кислоту \(\ce{H3PO3}\) (рис. 18.53). Лучший способ получения чистой фосфористой кислоты — гидролиз трихлорида фосфора:
Нагреванием полученного раствора удаляют хлороводород и выпаривают воду. Когда испарится достаточно воды, при охлаждении выпадают белые кристаллы фосфористой кислоты. Кристаллы расплывающиеся, очень хорошо растворимы в воде и имеют запах, напоминающий чесночный. Твёрдое вещество плавится при \(70{,}1\ \text{°C}\) и около \(200\ \text{°C}\) разлагается путём диспропорционирования на фосфин и ортофосфорную кислоту:
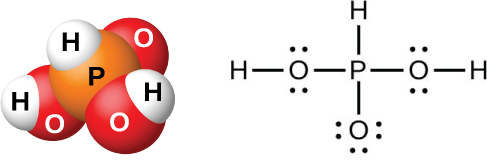
Рис. 18.53. В молекуле фосфористой кислоты \(\ce{H3PO3}\) только два атома водорода, связанные с атомом кислорода, обладают кислотными свойствами.
Фосфористая кислота образует лишь два ряда солей, содержащих дигидрофосфит-ион \(\ce{H2PO3-}\) или гидрофосфит-ион \(\ce{HPO3^{2-}}\) соответственно. Третий атом водорода заместить нельзя, поскольку он малокислотный: связь \(\ce{P-H}\) не ионизируется.
Кислородные кислоты серы и их соли¶
Получение серной кислоты \(\ce{H2SO4}\) (рис. 18.54) начинают с окисления серы до триоксида серы, а затем триоксид превращают в серную кислоту. Чистая серная кислота — бесцветная маслянистая жидкость, замерзающая при \(10{,}5\ \text{°C}\). При нагревании она дымит, поскольку разлагается на воду и триоксид серы. При нагревании теряется больше триоксида серы, чем воды, до тех пор, пока концентрация не достигнет 98,33 %. Кислота этой концентрации кипит при \(338\ \text{°C}\) без дальнейшего изменения концентрации (постоянно кипящий раствор), и это и есть коммерчески доступная концентрированная \(\ce{H2SO4}\). По количеству производимой серной кислоты промышленность опережает любое другое соединение.
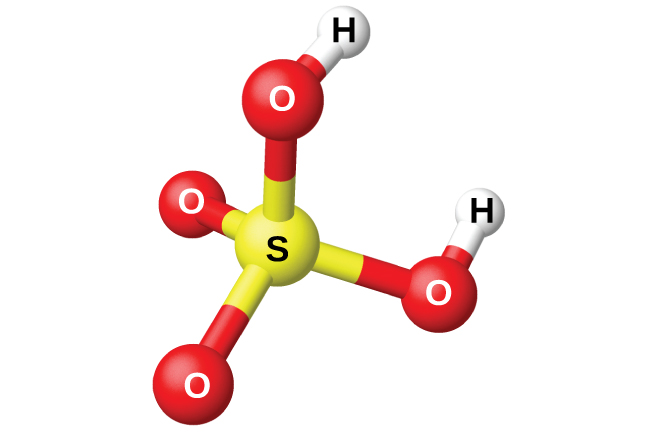
Рис. 18.54. Серная кислота имеет тетраэдрическое молекулярное строение.
Сильное сродство концентрированной серной кислоты к воде делает её хорошим осушителем (dehydrating agent). Газы и несмешивающиеся с кислотой жидкости, не реагирующие с ней, можно осушать, пропуская их через серную кислоту.
Серная кислота — сильная двухосновная кислота, ионизирующаяся в две ступени. В водном растворе первая ступень ионизации практически полная. Вторая ступень полная далеко не настолько, и ион \(\ce{HSO4-}\) — кислота умеренной силы (около 25 % ионизировано в растворе соли \(\ce{HSO4-}\): \(K_a = 1{,}2 \times 10^{-2}\)).
Будучи двухосновной кислотой, серная кислота образует и сульфаты, такие как \(\ce{Na2SO4}\), и гидросульфаты, такие как \(\ce{NaHSO4}\). Большинство сульфатов растворимы в воде; однако сульфаты бария, стронция, кальция и свинца лишь немного растворимы.
К важным сульфатам относятся \(\ce{Na2SO4 \cdot 10H2O}\) и английская соль (Epsom salts) \(\ce{MgSO4 \cdot 7H2O}\). Поскольку ион \(\ce{HSO4-}\) — кислота, гидросульфаты, такие как \(\ce{NaHSO4}\), проявляют кислотные свойства, и это соединение — основной компонент некоторых бытовых чистящих средств.
Горячая концентрированная серная кислота — окислитель. В зависимости от концентрации, температуры и силы восстановителя серная кислота окисляет многие соединения, восстанавливаясь при этом до \(\ce{SO2}\), \(\ce{HSO3-}\), \(\ce{SO3^{2-}}\), \(\ce{S}\), \(\ce{H2S}\) или \(\ce{S^{2-}}\).
Диоксид серы растворяется в воде, образуя раствор сернистой кислоты, как и подобает оксиду неметалла. Сернистая кислота неустойчива, и выделить безводную \(\ce{H2SO3}\) невозможно. При нагревании раствора сернистой кислоты диоксид серы улетучивается. Как и другие двухосновные кислоты, сернистая кислота ионизируется в две ступени: образуются гидросульфит-ион \(\ce{HSO3-}\) и сульфит-ион \(\ce{SO3^{2-}}\). Сернистая кислота — кислота умеренной силы. На первой ступени ионизация составляет около 25 %, а на второй — гораздо меньше (\(K_{a1} = 1{,}2 \times 10^{-2}\) и \(K_{a2} = 6{,}2 \times 10^{-8}\)).
Для получения твёрдых сульфитов и гидросульфитов к раствору сернистой кислоты добавляют стехиометрическое количество основания и затем выпаривают воду. Эти соли образуются также при реакции \(\ce{SO2}\) с оксидами и гидроксидами. При нагревании твёрдого гидросульфита натрия образуются сульфит натрия, диоксид серы и вода:
Сильные окислители способны окислять сернистую кислоту. Кислород воздуха медленно окисляет её до более устойчивой серной кислоты:
Растворы сульфитов также легко окисляются на воздухе с образованием сульфатов. Поэтому в растворах сульфитов после контакта с воздухом всегда содержатся и сульфаты.
Кислородные кислоты галогенов и их соли¶
Соединения \(\ce{HXO}\), \(\ce{HXO2}\), \(\ce{HXO3}\) и \(\ce{HXO4}\), где \(X\) — это \(\ce{Cl}\), \(\ce{Br}\) или \(\ce{I}\), — это соответственно галогеноватистые (hypohalous), галогенистые (halous), галогеноватые (halic) и галогенные (perhalic) кислоты. Сила этих кислот возрастает от галогеноватистых, которые очень слабы, до галогенных, которые очень сильны. Таблица 18.2 перечисляет известные кислоты с указанием их значений \(\mathrm{p}K_a\) в скобках там, где они известны.
| Название | Фтор | Хлор | Бром | Иод |
|---|---|---|---|---|
| галогеноватистая (hypohalous) | \(\ce{HOF}\) | \(\ce{HOCl}\) (7,5) | \(\ce{HOBr}\) (8,7) | \(\ce{HOI}\) (11) |
| галогенистая (halous) | \(\ce{HClO2}\) (2,0) | |||
| галогеноватая (halic) | \(\ce{HClO3}\) | \(\ce{HBrO3}\) | \(\ce{HIO3}\) (0,8) | |
| галогенная (perhalic) | \(\ce{HClO4}\) | \(\ce{HBrO4}\) | \(\ce{HIO4}\) (1,6) | |
| парагалогенная (paraperhalic) | \(\ce{H5IO6}\) (1,6) |
Таблица 18.2. Оксикислоты галогенов.
Единственная известная оксикислота фтора — очень неустойчивая фтороватистая кислота \(\ce{HOF}\), которую получают реакцией газообразного фтора со льдом:
Соединение очень неустойчиво и выше \(-40\ \text{°C}\) разлагается. Оно не ионизируется в воде, и его солей не известно. Не вполне ясно, уместно ли вообще название «фтороватистая кислота» для \(\ce{HOF}\); более подходящим названием может быть «гипофторит водорода».
Реакции хлора и брома с водой аналогичны реакции фтора со льдом, но они не идут до конца, и получаются смеси галогена с соответствующими галогеноватистыми и галогеноводородными кислотами. Кроме \(\ce{HOF}\), галогеноватистые кислоты существуют только в растворе. Все они очень слабые кислоты; однако \(\ce{HOCl}\) — более сильная, чем \(\ce{HOBr}\), а \(\ce{HOBr}\), в свою очередь, сильнее \(\ce{HOI}\).
Добавление основания к растворам галогеноватистых кислот даёт растворы солей, содержащих основные гипогалит-ионы \(\ce{OX-}\). Эти соли можно выделить в твёрдом виде. Все гипогалиты в растворе неустойчивы относительно диспропорционирования, но для гипохлорита реакция идёт медленно. Гипобромит и гипоиодит диспропорционируют быстро даже на холоду:
Гипохлорит натрия — недорогой отбеливатель (бытовое название Clorox) и бактерицид. Промышленно его получают электролизом холодных разбавленных водных растворов хлорида натрия в условиях, когда образующиеся хлор и гидроксид-ионы могут реагировать. Суммарная реакция:
Единственная определённо известная галогенистая кислота — хлористая \(\ce{HClO2}\), получаемая взаимодействием хлорита бария с разбавленной серной кислотой:
Отфильтровывание нерастворимого сульфата бария оставляет раствор \(\ce{HClO2}\). Хлористая кислота неустойчива; в растворе она медленно разлагается с образованием диоксида хлора, соляной кислоты и воды. Хлористая кислота реагирует с основаниями, образуя соли, содержащие хлорит-ион (рис. 18.55). Хлорит натрия находит широкое применение в отбеливании бумаги, так как он — сильный окислитель и не повреждает бумагу.
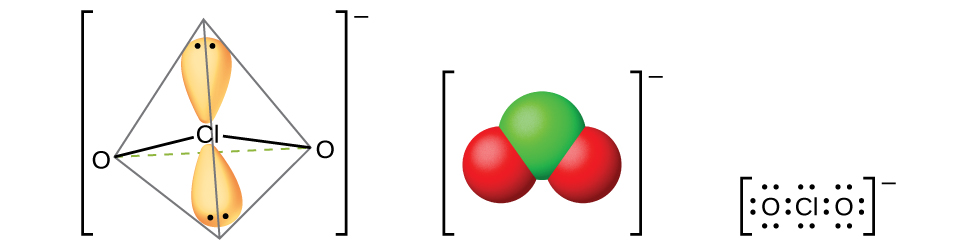
Рис. 18.55. Хлорит-ионы \(\ce{ClO2-}\) образуются при взаимодействии хлористой кислоты с основаниями.
Хлорноватая кислота \(\ce{HClO3}\) и бромноватая кислота \(\ce{HBrO3}\) устойчивы только в растворе. При взаимодействии иода с концентрированной азотной кислотой получают устойчивую белую иодноватую кислоту \(\ce{HIO3}\):
Более лёгкие галогеноватые кислоты можно получить из их бариевых солей реакцией с разбавленной серной кислотой. Реакция аналогична той, которой получают хлористую кислоту. Все галогеноватые кислоты — сильные кислоты и весьма активные окислители. Эти кислоты реагируют с основаниями, образуя соли, содержащие хлорат-ионы (рис. 18.56). Другой препаративный метод — электрохимическое окисление горячего раствора галогенида металла до соответствующего хлората металла. Хлорат натрия — гербицид; хлорат калия применяют как окислитель.
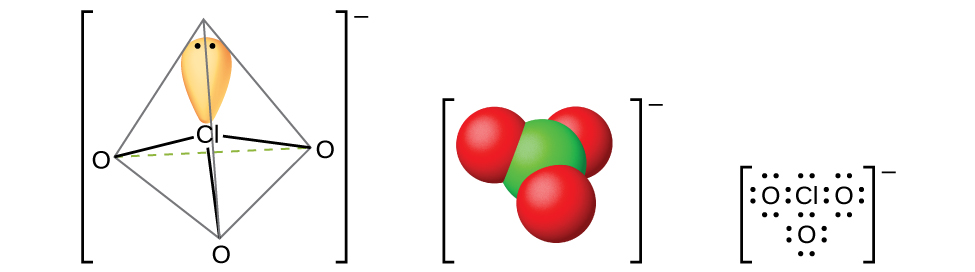
Рис. 18.56. Хлорат-ионы \(\ce{ClO3-}\) образуются при взаимодействии галогеноватых кислот с основаниями.
Хлорную кислоту \(\ce{HClO4}\) получают, обрабатывая перхлорат, например перхлорат калия, серной кислотой при пониженном давлении. \(\ce{HClO4}\) можно отогнать из смеси:
Разбавленные водные растворы хлорной кислоты вполне термически устойчивы, однако концентрации выше 60 % неустойчивы и опасны. Хлорная кислота и её соли — сильные окислители, поскольку очень электроотрицательному хлору устойчивее находиться в более низкой степени окисления, чем \(+7\). Известны случаи серьёзных взрывов при нагревании концентрированных растворов с легко окисляющимися веществами. Однако в холодном разбавленном виде реакции хлорной кислоты как окислителя идут медленно. Эта кислота — одна из самых сильных кислот вообще. Большинство солей, содержащих перхлорат-ион (рис. 18.57), растворимы. Их можно получить реакцией оснований с хлорной кислотой, а в промышленности — электролизом горячих растворов соответствующих хлоридов.
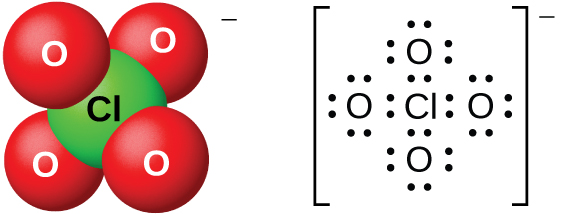
Рис. 18.57. Перхлорат-ионы \(\ce{ClO4-}\) образуются при взаимодействии хлорной кислоты с основаниями или электролизом горячих растворов хлоридов.
Соли пербромной кислоты получить трудно; на сегодня лучший способ их синтеза — окисление броматов в щелочном растворе газообразным фтором с последующим подкислением. Эта кислота и её соли коммерческого применения практически не имеют.
Существует несколько различных кислот, содержащих иод в степени окисления \(+7\); к ним относятся метапериодическая кислота \(\ce{HIO4}\) и парапериодическая кислота \(\ce{H5IO6}\). Эти кислоты — сильные окислители и реагируют с основаниями, образуя соответствующие соли.