17.7 Катализ (Catalysis)¶
Цели обучения
К концу этого раздела вы сможете:
- объяснять функцию катализатора в терминах механизмов реакций и диаграмм потенциальной энергии;
- приводить примеры катализа в природных и промышленных процессах.
Катализаторы не влияют на равновесие¶
Катализатор способен увеличить скорость реакции. Хотя такое ускорение может привести к тому, что система быстрее достигает равновесия (за счёт одновременного ускорения прямой и обратной реакций), катализатор не влияет ни на значение константы равновесия, ни на равновесные концентрации.
Взаимодействие изменений концентрации (или давления), температуры и отсутствие влияния катализатора на химическое равновесие хорошо иллюстрируется промышленным синтезом аммиака из азота и водорода по уравнению
Это одна из реакций, в которой производится огромное количество аммиака. Ежегодно аммиак входит в первую десятку химических веществ мира по массе производства. В США каждый год производится около 2 миллиардов фунтов (около 0,9 млн т) аммиака.
Аммиак играет важнейшую роль в мировой экономике. Он используется для производства удобрений и сам является важным удобрением для роста кукурузы, хлопчатника и других культур. Большие количества аммиака превращают в азотную кислоту, которая, в свою очередь, идёт на производство удобрений, взрывчатых веществ, пластмасс, красителей и волокон, а также применяется в металлургии.
Портрет химика. Фриц Габер (Fritz Haber)
В начале XX века немецкий химик Фриц Габер (Fritz Haber; рис. 17.19) разработал практический способ превращения двухатомного азота — в той форме, в какой его не могут усваивать растения, — в аммиак, форму азота, наиболее доступную растениям для усвоения.
Доступность азота — один из основных лимитирующих факторов роста растений. Несмотря на то что азот составляет 78 % воздуха, двухатомный азот (\(\ce{N2}\)) биологически недоступен из-за чрезвычайной прочности тройной связи азот–азот. Чтобы растения могли использовать атмосферный азот, его нужно перевести в более биологически доступную форму; это превращение называют связыванием азота (nitrogen fixation).
Габер родился в Бреслау, Пруссия (ныне Вроцлав, Польша), в декабре 1868 года. Он изучал химию и, работая в Технологическом университете Карлсруэ, разработал то, что впоследствии стало известно как процесс Габера (Haber process): каталитическое получение аммиака из водорода и атмосферного азота при высоких температурах и давлениях. За эту работу Габер был удостоен Нобелевской премии по химии 1918 года за синтез аммиака из элементов. Процесс Габера стал благом для сельского хозяйства, так как позволил производству удобрений больше не зависеть от добываемого сырья, такого как нитрат натрия. В настоящее время ежегодное производство синтетических азотных удобрений превышает 100 миллионов тонн, и благодаря синтетическим удобрениям число людей, которых способна прокормить единица пахотных земель, выросло с 1,9 человек на гектар в 1908 году до 4,3 в 2008 году.

Рис. 17.19. Работа лауреата Нобелевской премии Фрица Габера произвела революцию в сельскохозяйственной практике начала XX века. Его деятельность также повлияла на стратегию военных действий, добавив к артиллерии химическое оружие.
Помимо работ в области производства аммиака, Габер известен в истории и как один из «отцов» химической войны. Во время Первой мировой войны он сыграл важную роль в разработке отравляющих газов, применявшихся в окопной войне. О своей роли в этих разработках Габер говорил: «В мирное время учёный принадлежит миру, а в военное — своей стране»1. Габер защищал применение химического оружия от обвинений в бесчеловечности, говоря, что смерть есть смерть, каким бы способом она ни была причинена. Его судьба служит примером этических дилемм, стоящих перед учёными в военное время, и двойственной природы «меча науки».
Подобно самому Габеру, продукты, получаемые из аммиака, многолики. Помимо ценности для сельского хозяйства, азотсодержащие соединения могут служить и разрушительным целям. Нитрат аммония также использовался в составе взрывчатых веществ, в том числе самодельных взрывных устройств. Нитрат аммония был одним из компонентов бомбы, применённой при нападении на федеральное здание имени Альфреда П. Мурра в центре Оклахома-Сити 19 апреля 1995 года.
Давно известно, что азот и водород реагируют с образованием аммиака. Однако наладить производство аммиака в практически значимых количествах реакцией азота и водорода стало возможным лишь в начале XX века — после того, как были изучены факторы, влияющие на положение этого равновесия.
Чтобы промышленный процесс был экономически оправдан, он должен давать большой выход продукта за разумное время. Один из способов повысить выход аммиака — увеличить давление в системе, в которой \(\ce{N2}\), \(\ce{H2}\) и \(\ce{NH3}\) находятся в равновесии или приближаются к нему:
Образование дополнительного количества аммиака уменьшает общее давление, создаваемое системой, и тем самым отчасти ослабляет действие возросшего внешнего давления.
Хотя повышение давления смеси \(\ce{N2}\), \(\ce{H2}\) и \(\ce{NH3}\) увеличивает выход аммиака, при низких температурах скорость образования аммиака мала. При комнатной температуре, например, реакция настолько медленна, что если бы мы приготовили смесь \(\ce{N2}\) и \(\ce{H2}\), заметного количества аммиака не образовалось бы за всю нашу жизнь. Образование аммиака из водорода и азота — экзотермический процесс:
Поэтому повышение температуры, увеличивающее скорость, снижает выход. Если же мы понизим температуру, чтобы сдвинуть равновесие в сторону образования аммиака, то равновесие будет достигаться медленнее из-за резкого уменьшения скорости реакции с понижением температуры.
Часть скорости образования, теряемой при работе при более низких температурах, можно восполнить применением катализатора. Суммарное действие катализатора на реакцию сводится к тому, что равновесие достигается быстрее.
В промышленном производстве аммиака применяют условия около \(500\ \text{°C}\), \(150{-}900\ \text{атм}\) и присутствие катализатора, что даёт наилучший компромисс между скоростью, выходом и стоимостью оборудования, необходимого для получения и удержания газов при высоких давлениях и температурах (рис. 17.20).
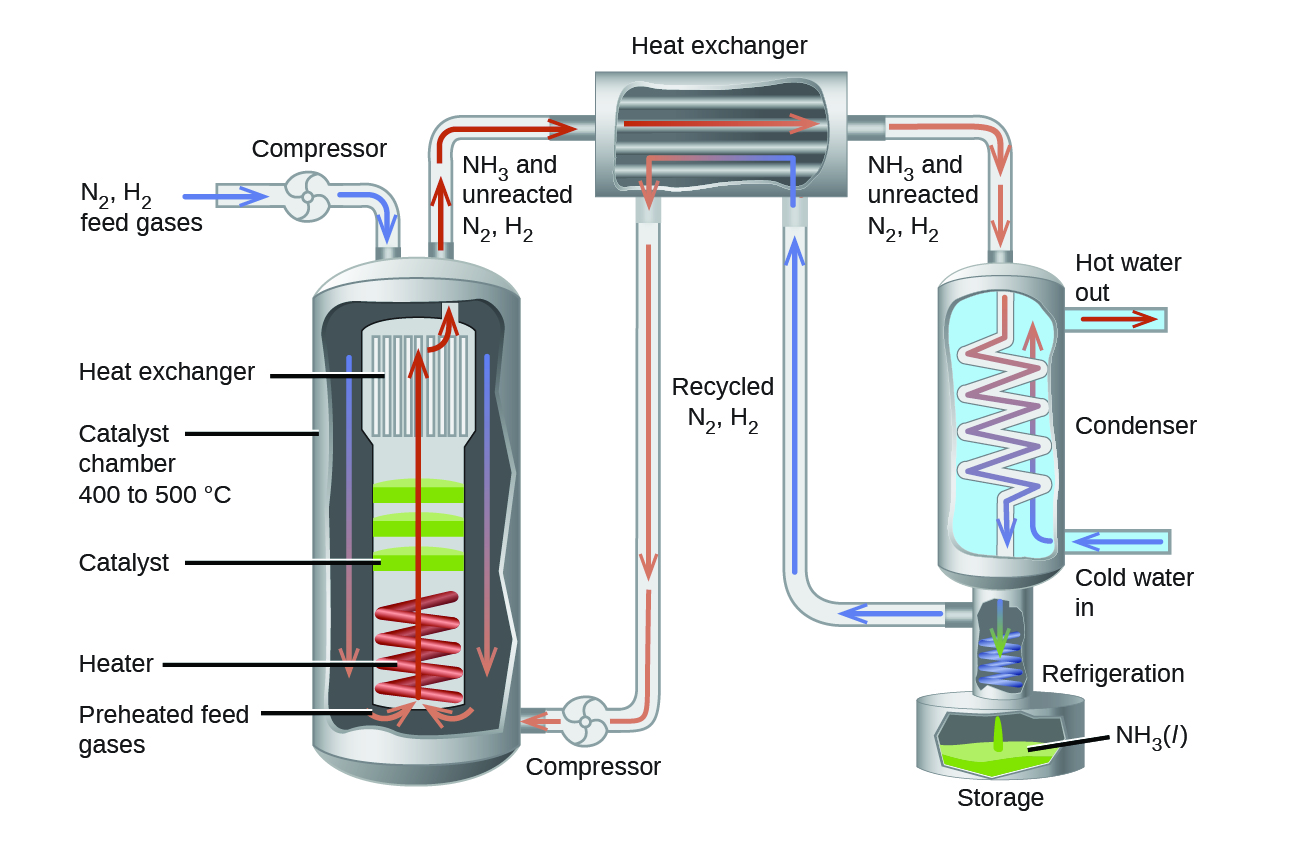
Рис. 17.20. Промышленное производство аммиака требует тяжёлого оборудования, способного работать при высоких температурах и давлениях. На схеме изображено устройство аммиачного завода.
Среди факторов, влияющих на скорость химических реакций и обсуждавшихся ранее в этой главе, упоминалось присутствие катализатора (catalyst) — вещества, способного увеличивать скорость реакции, не расходуясь в ней. Понятия, введённые в предыдущем разделе о механизмах реакций, дают основу для понимания того, как именно катализаторы выполняют эту важнейшую функцию.
На рис. 17.21 показаны диаграммы реакции для химического процесса в отсутствие и в присутствии катализатора. Рассмотрение этих диаграмм выявляет несколько характерных черт. В соответствии с тем, что обе диаграммы изображают одну и ту же суммарную реакцию, обе кривые начинаются и заканчиваются при одних и тех же энергиях (в данном случае, поскольку продукты обладают более высокой энергией, чем реагенты, реакция эндотермическая). Однако механизмы реакций явно различны. Некаталитическая реакция протекает по одностадийному механизму (наблюдается одно переходное состояние), тогда как каталитическая реакция следует двустадийному механизму (два переходных состояния) с заметно меньшей энергией активации. Это различие иллюстрирует способ, которым катализатор ускоряет реакции, а именно: он предоставляет альтернативный путь реакции с меньшей энергией активации. Хотя каталитический механизм реакции не обязательно должен включать иное число стадий, чем некаталитический, он должен обеспечивать путь, у которого лимитирующая стадия идёт быстрее (меньше \(E_a\)).
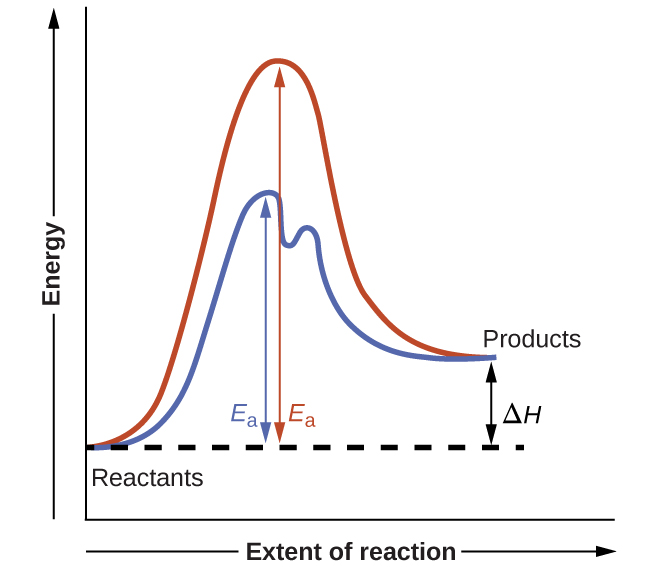
Рис. 17.21. Диаграммы реакции для эндотермического процесса в отсутствие (красная кривая) и в присутствии (синяя кривая) катализатора. Каталитический путь включает двустадийный механизм (обратите внимание на два переходных состояния) и промежуточное вещество (изображено впадиной между двумя переходными состояниями).
Пример 17.15. Диаграммы реакции для каталитических реакций
Задача. Две приведённые ниже диаграммы реакции изображают одну и ту же реакцию: одну без катализатора, другую — с катализатором. Оцените энергию активации для каждого процесса и определите, какая из диаграмм соответствует каталитической реакции.
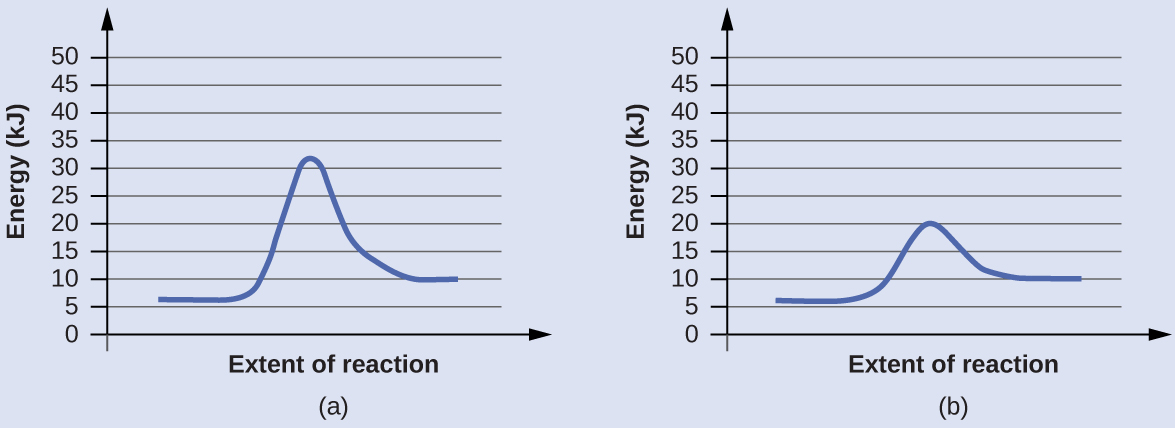
Решение. Энергии активации рассчитываются как разность энергии переходного состояния и энергии реагентов:
Каталитической является реакция с меньшей энергией активации; в данном случае ей соответствует диаграмма (b).
Проверь себя. Ниже показаны диаграммы реакции для химического процесса с катализатором и без него. Обе реакции включают двустадийный механизм с лимитирующей первой стадией. Рассчитайте энергии активации для первой стадии каждого механизма и определите, какая из них соответствует каталитической реакции. Сравните вторые стадии двух механизмов.
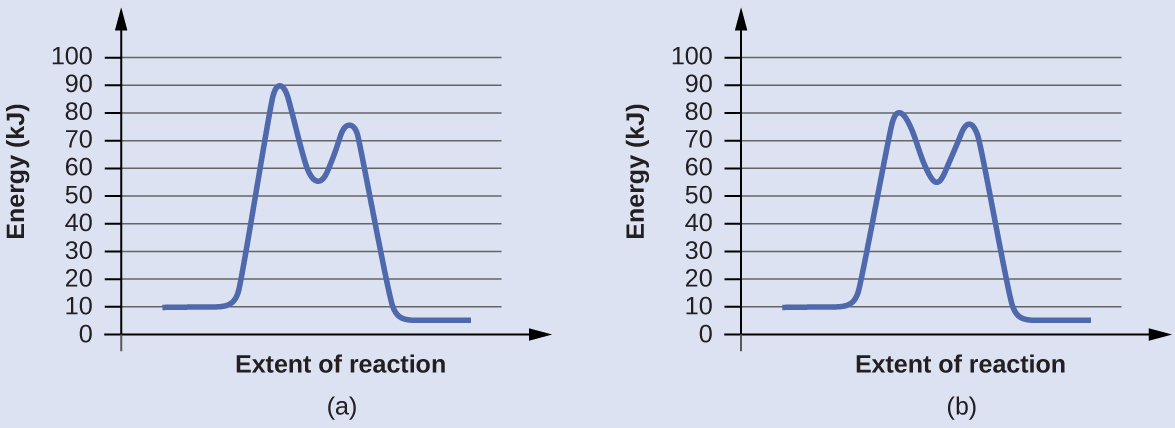
Ответ. Для первой стадии \(E_a = 80\ \text{кДж}\) для (a) и \(70\ \text{кДж}\) для (b), поэтому каталитической реакции соответствует диаграмма (b). Энергии активации вторых стадий обоих механизмов одинаковы и равны \(20\ \text{кДж}\).
Гомогенные катализаторы¶
Гомогенный катализатор (homogeneous catalyst) находится в той же фазе, что и реагенты. Он взаимодействует с реагентом, образуя промежуточное вещество, которое затем разлагается или реагирует с другим реагентом за одну или несколько стадий, регенерируя исходный катализатор и образуя продукт.
В качестве важной иллюстрации гомогенного катализа рассмотрим озоновый слой Земли. Озон в верхних слоях атмосферы, защищающий Землю от ультрафиолетового излучения, образуется при поглощении молекулами кислорода ультрафиолетового света и протекании реакции:
Озон — относительно неустойчивая молекула, разлагающаяся с образованием двухатомного кислорода по обратной реакции. Этот процесс разложения согласуется со следующим двустадийным механизмом:
Ряд веществ может катализировать разложение озона. Например, считается, что катализируемое оксидом азота(II) разложение озона происходит по следующему трёхстадийному механизму:
Как и должно быть, суммарная реакция одна и та же как для двустадийного некаталитического, так и для трёхстадийного механизма, катализируемого \(\ce{NO}\):
Обратите внимание, что \(\ce{NO}\) является реагентом на первой стадии механизма и продуктом — на последней. Это ещё одна характерная черта катализатора: участвуя в химической реакции, он не расходуется в ней.
Портрет химика. Марио Х. Молина (Mario J. Molina)
Нобелевская премия по химии 1995 года была разделена между Паулем Й. Крутценом, Марио Х. Молиной (рис. 17.22) и Ф. Шервудом Роулендом «за работы в области химии атмосферы, особенно касающиеся образования и разложения озона»2. Молина, гражданин Мексики, выполнил большую часть своих работ в Массачусетском технологическом институте (MIT).
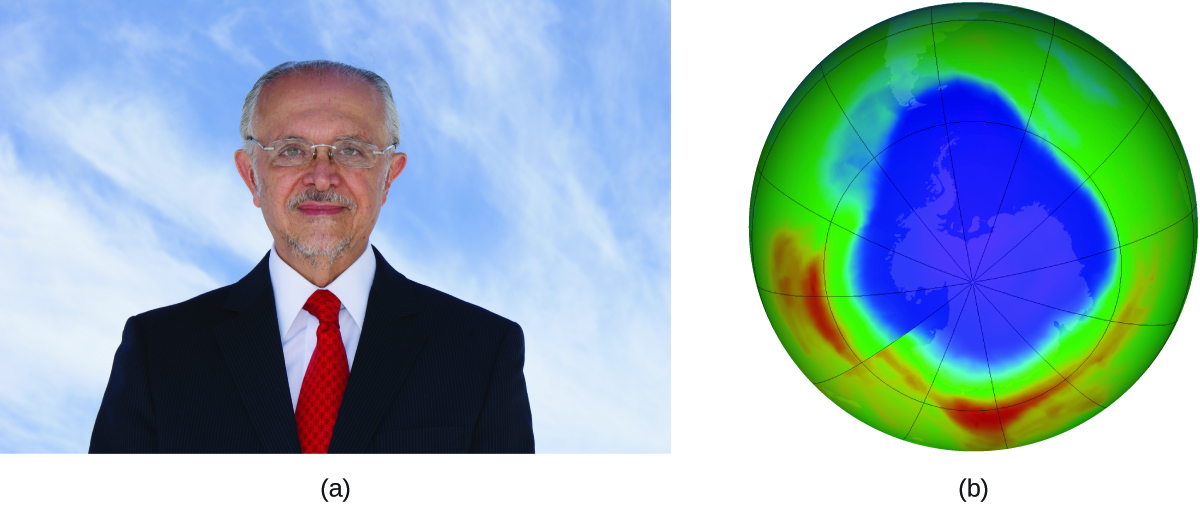
Рис. 17.22. (а) Мексиканский химик Марио Молина (1943–) разделил Нобелевскую премию по химии 1995 года за свои исследования (б) антарктической озоновой дыры. (credit a: courtesy of Mario Molina; credit b: modification of work by NASA)
В 1974 году Молина и Роуленд опубликовали в журнале Nature статью, в которой подробно описывалась угроза, представляемая хлорфторуглеродными газами для устойчивости озонового слоя в верхней атмосфере Земли. Озоновый слой защищает Землю от солнечной радиации, поглощая ультрафиолетовое излучение. По мере того как химические реакции истощают количество озона в верхней атмосфере, над Антарктидой образуется измеримая «дыра», и до поверхности Земли доходит больше солнечной ультрафиолетовой радиации, тесно связанной с распространённостью рака кожи. Работа Молины и Роуленда сыграла решающую роль в принятии Монреальского протокола — международного договора, подписанного в 1987 году, который успешно начал постепенное прекращение производства веществ, разрушающих озоновый слой.
Молина и Роуленд показали, что атомы хлора, выделяющиеся из созданных человеком веществ, способны катализировать разрушение озона в процессе, аналогичном тому, в котором \(\ce{NO}\) ускоряет разложение озона. Атомы хлора образуются, когда хлоруглероды или хлорфторуглероды — некогда широко применявшиеся в качестве хладагентов и пропеллентов — фотохимически разлагаются под действием ультрафиолетового света или реагируют с гидроксильными радикалами. Один из возможных механизмов показан ниже на примере метилхлорида:
Радикалы хлора разрушают озон и регенерируются в следующем каталитическом цикле:
Один одноатомный атом хлора способен разрушить тысячи молекул озона. К счастью, большая часть атмосферного хлора находится в каталитически неактивных формах — \(\ce{Cl2}\) и \(\ce{ClONO2}\).
После получения своей доли Нобелевской премии Молина продолжил работу в области химии атмосферы в MIT.
Связи между науками. Дефицит глюкозо-6-фосфатдегидрогеназы
Ферменты (enzymes) в организме человека действуют как катализаторы для важных химических реакций клеточного метаболизма. Поэтому недостаток того или иного фермента может приводить к опасной для жизни болезни. Дефицит G6PD (glucose-6-phosphate dehydrogenase, глюкозо-6-фосфатдегидрогеназы) — генетическое заболевание, выражающееся в нехватке фермента глюкозо-6-фосфатдегидрогеназы, — является самым распространённым ферментным дефицитом у людей. Этот фермент, показанный на рис. 17.23, представляет собой лимитирующий по скорости фермент метаболического пути, поставляющего клеткам \(\ce{NADPH}\) (рис. 17.24).
Рис. 17.23. Глюкозо-6-фосфатдегидрогеназа — лимитирующий по скорости фермент метаболического пути, поставляющего клеткам \(\ce{NADPH}\).
Нарушение этого пути может приводить к снижению уровня глутатиона в эритроцитах; как только весь глутатион израсходован, ферменты и другие белки, такие как гемоглобин, становятся уязвимыми для повреждения. Например, гемоглобин может превращаться в билирубин, что ведёт к желтухе — состоянию, способному стать тяжёлым. Люди, страдающие дефицитом G6PD, должны избегать определённых продуктов питания и лекарств, содержащих вещества, способные повреждать их обеднённые глутатионом эритроциты.
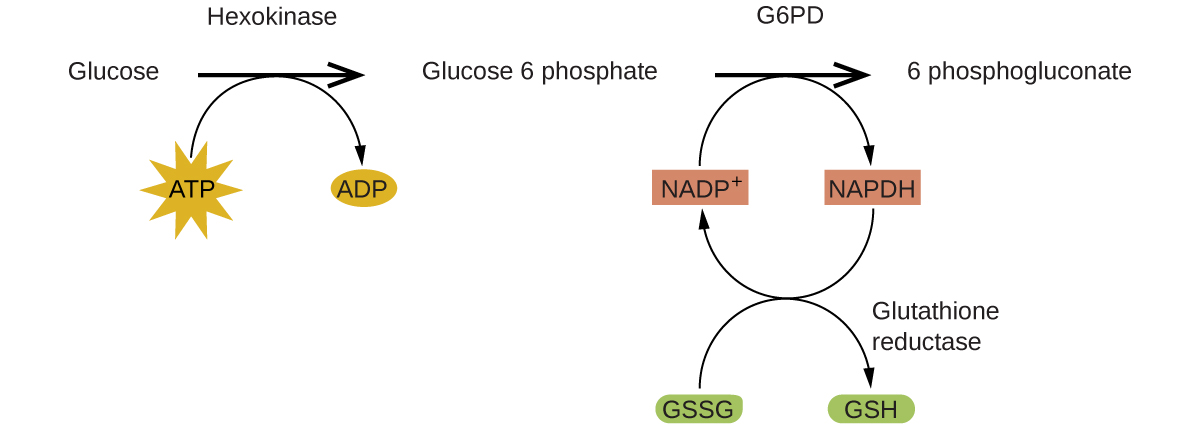
Рис. 17.24. В механизме пентозофосфатного пути G6PD катализирует реакцию, регулирующую \(\ce{NADPH}\) — кофермент, регулирующий глутатион, антиоксидант, защищающий эритроциты и другие клетки от окислительных повреждений.
Гетерогенные катализаторы¶
Гетерогенный катализатор (heterogeneous catalyst) — катализатор, находящийся в иной фазе (обычно в твёрдой), чем реагенты. Такие катализаторы, как правило, действуют, предоставляя активную поверхность, на которой может протекать реакция. Реакции в газовой и жидкой фазах, катализируемые гетерогенными катализаторами, происходят на поверхности катализатора, а не в объёме газа или жидкости.
Гетерогенный катализ обычно включает следующие процессы:
- Адсорбция (adsorption) реагента (или реагентов) на поверхности катализатора.
- Активация адсорбированного реагента (реагентов).
- Реакция адсорбированного реагента (реагентов).
- Десорбция (desorption) продукта (или продуктов) с поверхности катализатора.
На рис. 17.25 показаны стадии механизма реакции соединений с углерод-углеродной двойной связью с водородом на никелевом катализаторе. Никель — катализатор, используемый при гидрировании полиненасыщенных жиров и масел (содержащих несколько углерод-углеродных двойных связей) до насыщенных жиров и масел (содержащих только углерод-углеродные одинарные связи).
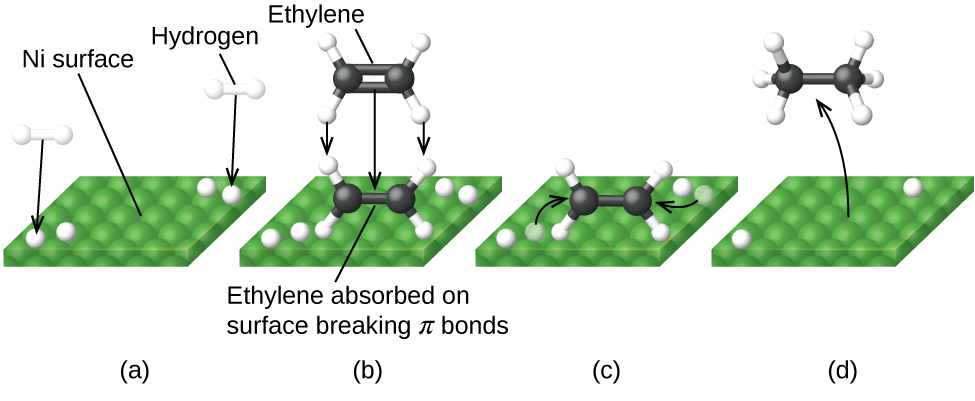
Рис. 17.25. Механизм катализируемой никелем реакции \(\ce{C2H4 + H2 -> C2H6}\). (а) Водород адсорбируется на поверхности, разрывая связи H–H и образуя связи Ni–H. (б) Этилен адсорбируется на поверхности, разрывая \(\pi\)-связь C–C и образуя связи Ni–C. (в) Атомы диффундируют по поверхности и образуют новые связи C–H при столкновениях. (г) Молекулы \(\ce{C2H6}\) десорбируются с никелевой поверхности.
Многие важные химические продукты получают в промышленности с применением гетерогенных катализаторов; к ним относятся аммиак, азотная кислота, серная кислота и метанол. Гетерогенные катализаторы используются и в каталитических нейтрализаторах (catalytic converters), устанавливаемых в большинстве автомобилей с бензиновыми двигателями (рис. 17.26).
Химия в повседневной жизни. Автомобильные каталитические нейтрализаторы
Учёные разработали каталитические нейтрализаторы для уменьшения количества токсичных выбросов, образующихся при сжигании бензина в двигателях внутреннего сгорания. Используя тщательно подобранную смесь каталитически активных металлов, удаётся одновременно осуществлять полное сгорание всех углеродсодержащих соединений до диоксида углерода и снижать выход оксидов азота. Это особенно удивительно, если учесть, что одна стадия предполагает присоединение к молекуле дополнительного кислорода, а другая — его удаление (рис. 17.26).
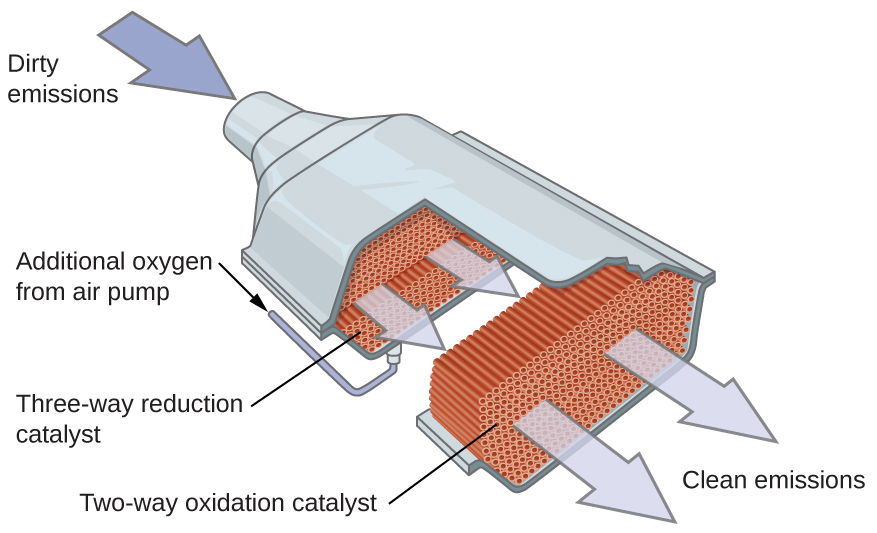
Рис. 17.26. Каталитический нейтрализатор обеспечивает сгорание всех углеродсодержащих соединений до диоксида углерода и одновременно снижает выбросы оксида азота и других загрязняющих веществ в выхлопе бензиновых двигателей.
Большинство современных трёхкомпонентных каталитических нейтрализаторов имеет поверхность, пропитанную платино-родиевым катализатором, который катализирует превращение оксида азота(II) в диазот и кислород, а также превращение оксида углерода(II) и углеводородов, таких как октан, в диоксид углерода и водяной пар:
Чтобы быть максимально эффективными, большинство каталитических нейтрализаторов предварительно подогреваются электрическим нагревателем. Это обеспечивает то, что металлы в катализаторе становятся полностью активными ещё до того, как выхлопные газы автомобиля прогреются достаточно, чтобы поддерживать нужные температуры реакции.
Дополнительно
«ChemWiki» Калифорнийского университета в Дейвисе предоставляет подробное описание того, как работают каталитические нейтрализаторы.
Связи между науками. Строение и функция ферментов
Изучение ферментов — важная точка соприкосновения биологии и химии. Ферменты обычно представляют собой белки (полипептиды), которые помогают регулировать скорость химических реакций между биологически важными соединениями, особенно теми, что участвуют в клеточном метаболизме. Разные классы ферментов выполняют различные функции, как показано в таблице 17.3.
Таблица 17.3. Классы ферментов и их функции
| Класс | Функция |
|---|---|
| оксидоредуктазы | окислительно-восстановительные реакции |
| трансферазы | перенос функциональных групп |
| гидролазы | реакции гидролиза |
| лиазы | отщепление группы с образованием двойной связи |
| изомеразы | изомеризация |
| лигазы | образование связи с гидролизом АТФ |
Молекулы ферментов имеют активный центр (active site) — часть молекулы такой формы, которая позволяет ей связываться с определённым субстратом (substrate) — молекулой-реагентом — с образованием фермент-субстратного комплекса (enzyme-substrate complex) в качестве промежуточного соединения реакции. Существуют две модели, объясняющие, как работает этот активный центр. Самая простая из них — модель «ключ – замок» (lock-and-key hypothesis), согласно которой формы активного центра и субстрата комплементарны и точно соответствуют друг другу, как ключ и замок. Гипотеза индуцированного соответствия (induced fit hypothesis), напротив, предполагает, что молекула фермента гибка и меняет свою форму, чтобы связаться с субстратом. Это не означает, что активный центр фермента полностью пластичен. И модель «ключ – замок», и модель индуцированного соответствия объясняют тот факт, что ферменты могут связываться только с определёнными субстратами, поскольку, как правило, конкретный фермент катализирует лишь конкретную реакцию (рис. 17.27).
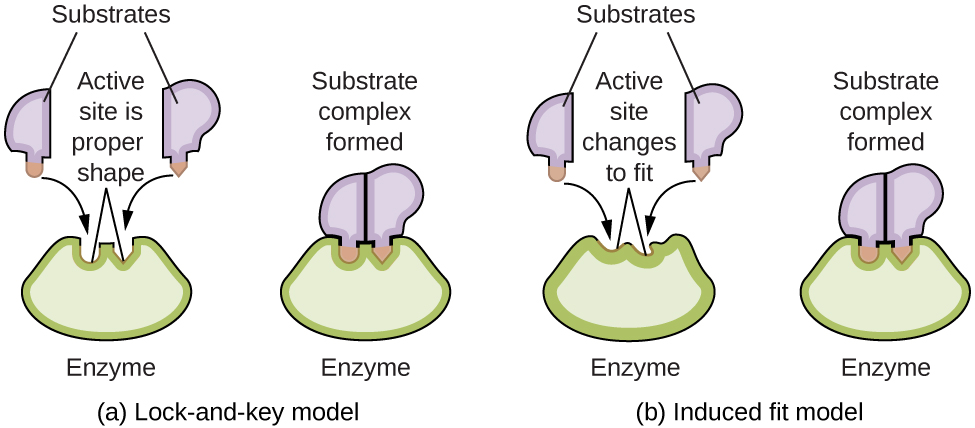
Рис. 17.27. (а) Согласно модели «ключ – замок», форма активного центра фермента идеально подходит для субстрата. (б) Согласно модели индуцированного соответствия, активный центр несколько гибок и может менять форму, чтобы связаться с субстратом.
Дополнительно
Королевское химическое общество предоставляет превосходное введение в ферменты для учащихся и преподавателей.
Связь между скоростью реакции и константой равновесия¶
Связь между скоростью реакции и её константой равновесия легко получить с помощью небольшой алгебраической подстановки. Для реакции, в которой вещество \(A\) превращается в \(B\) (и обратно),
скорость прямой реакции равна
а скорость обратной —
После установления равновесия скорости прямой и обратной реакций равны:
Произведя небольшое преобразование, получаем
Также вспомним, что константа равновесия равна отношению концентраций продуктов и реагентов в состоянии равновесия:
Таким образом, константа равновесия оказывается равной отношению констант скорости прямой и обратной реакций. Это соотношение также помогает закрепить понимание природы катализатора. Катализатор не изменяет фундаментального равновесия (как и лежащей в его основе термодинамики). Скорее, он изменяет константу скорости реакции — причём обе константы, прямую и обратную, одинаково. Тем самым катализаторы обычно ускоряют достижение равновесия (хотя могут использоваться и для замедления реакции!).
-
Herrlich, P. «The Responsibility of the Scientist: What Can History Teach Us About How Scientists Should Handle Research That Has the Potential to Create Harm?» EMBO Reports 14 (2013): 759–764. ↩
-
«The Nobel Prize in Chemistry 1995», Nobel Prize.org, дата обращения 18 февраля 2015 г., http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1995/. ↩