17.5 Теория столкновений (Collision Theory)¶
Цели обучения
К концу этого раздела вы сможете:
- использовать постулаты теории столкновений для объяснения влияния агрегатного состояния, температуры и концентрации на скорость реакции;
- сформулировать понятия энергии активации и переходного состояния;
- применять уравнение Аррениуса в расчётах, связывающих константу скорости с температурой.
Нет ничего удивительного в том, что атомы, молекулы или ионы должны столкнуться друг с другом, прежде чем смогут вступить в реакцию: чтобы образовались химические связи, атомы должны сблизиться. Это простое соображение лежит в основе мощной теории, объясняющей многочисленные наблюдения химической кинетики, в том числе и факторы, влияющие на скорость реакции.
Теория столкновений (collision theory) опирается на следующие постулаты:
-
Скорость реакции пропорциональна частоте столкновений исходных веществ:
\[ \text{скорость реакции} \propto \dfrac{\text{число столкновений}}{\text{время}} \] -
Реагирующие частицы должны столкнуться в такой ориентации, чтобы между атомами, которые в продукте окажутся связанными, был возможен контакт.
-
Столкновение должно произойти с энергией, достаточной для взаимного проникновения валентных оболочек реагирующих частиц, чтобы электроны могли перераспределиться и образовать новые связи (а значит, и новые химические частицы).
Важность двух физических факторов, отмеченных в постулатах 2 и 3, — ориентации и энергии столкновений — хорошо видна на примере реакции монооксида углерода с кислородом:
Монооксид углерода — загрязнитель, образующийся при сгорании углеводородного топлива. Для снижения его выбросов в автомобилях устанавливают каталитические нейтрализаторы, в которых катализатор обеспечивает протекание указанной реакции. Эта же реакция идёт как побочный процесс при сгорании пороха и даёт дульное пламя у многих видов огнестрельного оружия. При достаточном количестве монооксида углерода и кислорода реакция протекает при высокой температуре и давлении.
Первая стадия газофазной реакции между монооксидом углерода и кислородом — это столкновение двух молекул:
Хотя возможных взаимных ориентаций двух молекул много, рассмотрим две из них, показанные на рис. 17.13. В первом случае с молекулой кислорода сталкивается атом кислорода молекулы \(\ce{CO}\). Во втором случае с молекулой кислорода сталкивается атом углерода молекулы \(\ce{CO}\). Очевидно, что второй случай гораздо вероятнее приведёт к образованию диоксида углерода, в котором центральный атом углерода связан с двумя атомами кислорода (\(\ce{O=C=O}\)). Этот простой пример показывает, насколько важна ориентация столкновения для получения нужного продукта реакции.
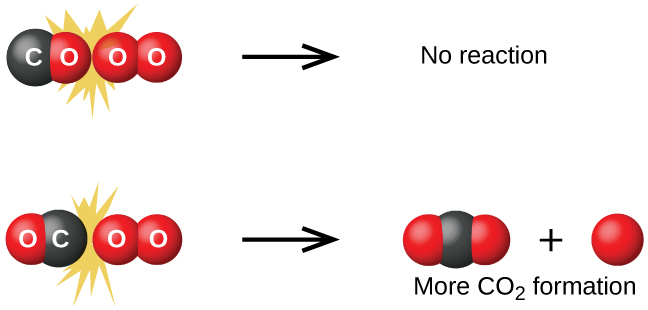
Рис. 17.13. Показаны два возможных варианта столкновения между молекулами монооксида углерода и кислорода. Ориентация сталкивающихся молекул отчасти определяет, произойдёт ли между ними реакция.
Даже если столкновение происходит с правильной ориентацией, это ещё не гарантирует образования диоксида углерода. Помимо подходящей ориентации, столкновение должно произойти и с достаточной энергией, чтобы образовался продукт. Когда реагирующие частицы сталкиваются и с правильной ориентацией, и с достаточной энергией, они объединяются в неустойчивую частицу, называемую активированным комплексом (activated complex), или переходным состоянием (transition state). Время жизни таких частиц очень мало, и большинство аналитических приборов их зарегистрировать не в состоянии; в отдельных случаях переходные состояния удавалось наблюдать тонкими спектральными методами.
Теория столкновений объясняет, почему скорость большинства реакций возрастает с увеличением концентрации. При увеличении концентрации любого из реагирующих веществ возрастает и вероятность столкновений между молекулами — на единицу объёма приходится больше молекул. Чем больше столкновений, тем выше скорость реакции — при условии, что их энергия достаточна.
Энергия активации и уравнение Аррениуса¶
Минимальная энергия, необходимая для образования продукта при столкновении реагентов, называется энергией активации (\(E_a\)) (activation energy). Соотношение этой энергии с кинетической энергией сталкивающихся молекул реагентов — главный фактор, определяющий скорость химической реакции. Если энергия активации существенно превышает среднюю кинетическую энергию молекул, реакция идёт медленно: достаточной для реакции энергией обладают лишь немногие быстрые молекулы. Если же энергия активации много меньше средней кинетической энергии молекул, большая их доля окажется достаточно энергичной, и реакция пойдёт быстро.
На рис. 17.14 показано, как меняется энергия химической системы в ходе превращения исходных веществ в продукты по схеме
Такие диаграммы реакции (reaction diagrams) широко используют в химической кинетике для иллюстрации различных свойств рассматриваемой реакции. Если читать диаграмму слева направо, в начальном состоянии система содержит только исходные вещества \(\ce{A} + \ce{B}\). Молекулы реагентов с достаточной энергией могут столкнуться и образовать высокоэнергичный активированный комплекс, или переходное состояние. Неустойчивое переходное состояние затем распадается с образованием стабильных продуктов \(\ce{C} + \ce{D}\). На диаграмме энергия активации реакции \(E_a\) изображена как разность энергий между исходными веществами и переходным состоянием. Если использовать конкретную форму энергии — энтальпию (см. главу о термохимии), — то изменение энтальпии реакции \(\Delta H\) оценивается как разность энергий между исходными веществами и продуктами. В данном случае реакция экзотермическая (\(\Delta H < 0\)), поскольку она сопровождается уменьшением энтальпии системы.
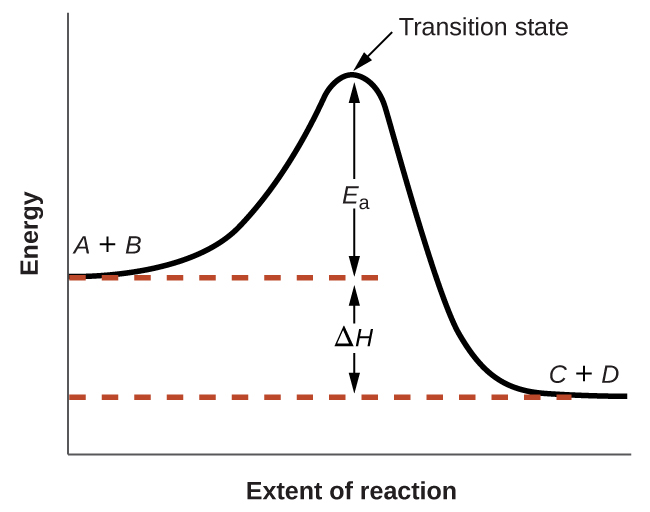
Рис. 17.14. Диаграмма экзотермической реакции \(\ce{A + B -> C + D}\).
Уравнение Аррениуса (Arrhenius equation) связывает энергию активации и константу скорости \(k\) для многих химических реакций:
В этом уравнении \(R\) — универсальная газовая постоянная, равная \(8{,}314\ \text{Дж}/(\text{моль}\cdot\text{К})\); \(T\) — температура по шкале Кельвина; \(E_a\) — энергия активации в джоулях на моль; \(e\) — постоянная, \(e = 2{,}7183\); \(A\) — постоянная, называемая предэкспоненциальным (частотным) множителем (frequency factor); она связана с частотой столкновений и их ориентацией.
Постулаты теории столкновений хорошо согласуются с уравнением Аррениуса. Предэкспоненциальный множитель \(A\) отражает, насколько условия реакции благоприятствуют правильно ориентированным столкновениям между молекулами реагентов. Чем выше вероятность эффективно ориентированных столкновений, тем больше \(A\) и тем выше скорость реакции.
Экспоненциальный сомножитель \(e^{-E_a/RT}\) описывает влияние энергии активации на скорость реакции. Согласно кинетико-молекулярной теории (см. главу о газах), температура вещества — это мера средней кинетической энергии составляющих его атомов или молекул. Распределение энергий между молекулами в образце вещества при заданной температуре описывается кривой на рис. 17.15(a). Две заштрихованные области под кривой соответствуют числу молекул, обладающих энергией (\(RT\)), достаточной для преодоления активационных барьеров (\(E_a\)). Меньшая энергия активации даёт большую долю достаточно энергичных молекул и более быструю реакцию.
Тот же экспоненциальный сомножитель описывает и влияние температуры на скорость реакции. Более высокой температуре отвечает большая доля молекул с энергией (\(RT\)), достаточной для преодоления активационного барьера (\(E_a\)), как показано на рис. 17.15(b). Это даёт большее значение константы скорости и, соответственно, большую скорость реакции.
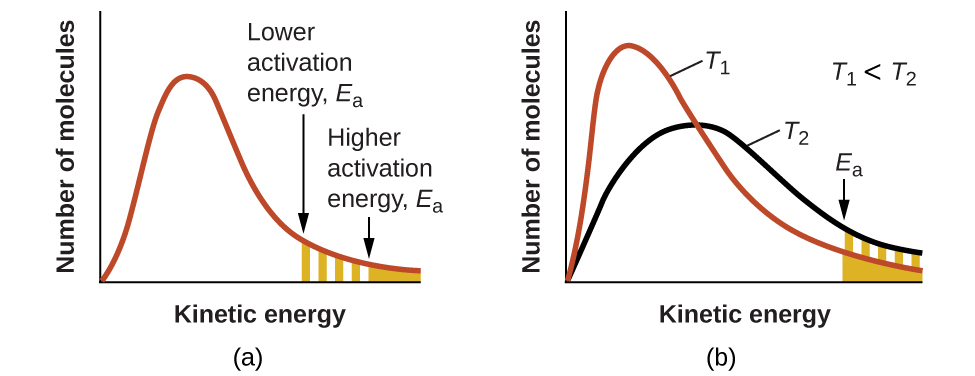
Рис. 17.15. Распределения молекул по энергиям, показывающие число молекул с энергией, превышающей (a) две разные энергии активации при заданной температуре и (b) заданную энергию активации при двух разных температурах.
Удобный способ определения \(E_a\) для реакции состоит в измерении \(k\) при двух или более температурах и использовании логарифмической формы уравнения Аррениуса, имеющей вид линейного уравнения:
График зависимости \(\ln k\) от \(\dfrac{1}{T}\) линеен; наклон прямой равен \(-\dfrac{E_a}{R}\), а отрезок, отсекаемый на оси ординат, равен \(\ln A\).
Пример 17.13. Определение \(E_a\)
Зависимость константы скорости от температуры для разложения \(\ce{HI(g)}\) на \(\ce{H2(g)}\) и \(\ce{I2(g)}\) приведена ниже. Какова энергия активации этой реакции?
| \(T\) (К) | \(k\) (л/моль/с) |
|---|---|
| 555 | \(3{,}52 \times 10^{-7}\) |
| 575 | \(1{,}22 \times 10^{-6}\) |
| 645 | \(8{,}59 \times 10^{-5}\) |
| 700 | \(1{,}16 \times 10^{-3}\) |
| 781 | \(3{,}95 \times 10^{-2}\) |
Решение. По исходным данным получим значения \(\dfrac{1}{T}\) и \(\ln k\):
| \(\dfrac{1}{T}\) (К\(^{-1}\)) | \(\ln k\) |
|---|---|
| \(1{,}80 \times 10^{-3}\) | \(-14{,}860\) |
| \(1{,}74 \times 10^{-3}\) | \(-13{,}617\) |
| \(1{,}55 \times 10^{-3}\) | \(-9{,}362\) |
| \(1{,}43 \times 10^{-3}\) | \(-6{,}759\) |
| \(1{,}28 \times 10^{-3}\) | \(-3{,}231\) |
На рис. 17.16 построен график \(\ln k\) от \(\dfrac{1}{T}\). На практике уравнение прямой (наклон и точку пересечения с осью), наиболее подходящей к этим точкам, получают статистической процедурой — регрессией. Это полезно для большинства экспериментальных данных, поскольку идеальное попадание всех точек на прямую встречается редко. Для приведённых данных согласие почти идеальное, и наклон можно оценить по любой паре точек. Возьмём первую и последнюю точки.
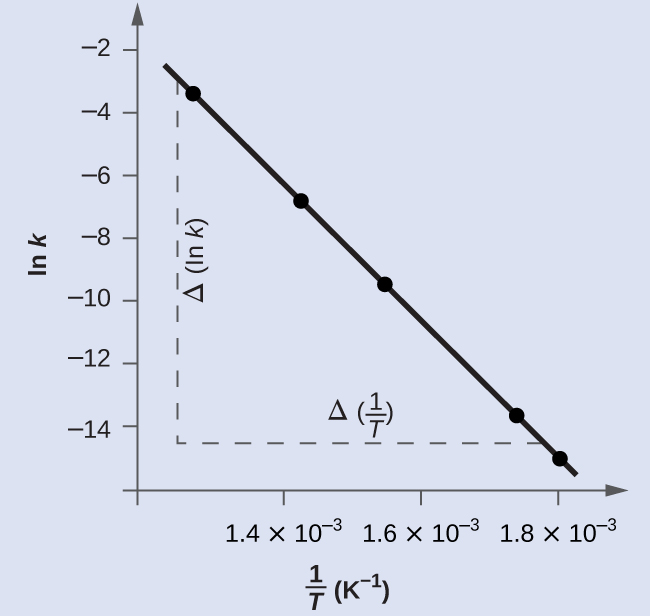
Рис. 17.16. График показывает линейную зависимость \(\ln k\) от \(\dfrac{1}{T}\) для реакции \(\ce{2HI(g) -> H2(g) + I2(g)}\) в соответствии с уравнением Аррениуса.
Альтернативный подход. Более быстрый способ — определить энергию активации по измерениям константы скорости всего при двух температурах. В этом подходе уравнение Аррениуса переписывают в удобной двухточечной форме:
Преобразуя это уравнение к виду, выражающему энергию активации, получаем:
В это уравнение можно подставить любую пару значений — например, первую и последнюю строки приведённой выше таблицы:
и получаем \(E_a = 1{,}8 \times 10^{5}\ \text{Дж}\,\text{моль}^{-1}\), или \(180\ \text{кДж}\,\text{моль}^{-1}\).
Этот способ даёт тот же результат, что и более строгий графический подход, как и следовало ожидать. На практике графический подход обычно даёт более надёжные результаты при работе с реальными экспериментальными данными.
Проверь себя. Константа скорости разложения \(\ce{N2O5}\) на \(\ce{NO}\) и \(\ce{O2}\) в газовой фазе равна \(1{,}66\ \text{л}/(\text{моль}\cdot\text{с})\) при \(650\ \text{К}\) и \(7{,}39\ \text{л}/(\text{моль}\cdot\text{с})\) при \(700\ \text{К}\):
В предположении, что кинетика этой реакции описывается уравнением Аррениуса, рассчитайте энергию активации этого разложения.
Ответ: \(1{,}1 \times 10^{5}\ \text{Дж}\,\text{моль}^{-1}\), или \(110\ \text{кДж}\,\text{моль}^{-1}\).